Introduction
About this Database
This database contains predictions of fibril-forming segments within proteins identified by the 3D Profile Method described below. We have analyzed over 20,000 putative protein sequences for segments with high fibrillation propensity that could form a "steric zipper"—two self-complementary beta sheets, giving rise to the spine of an amyloid fibril. Our approach is unique in that we use structural information to evaluate the likelihood that a particular sequence can form fibrils.
Prediction of Fibril-Forming Segments
Fibrillation propensities were computed using a structure-based algorithm originally described by Thompson et al., PNAS, 2006. This algorithm uses the crystal structure of the fibril-forming peptide NNQQNY from the sup35 prion protein of Saccharomyces cerevisiae which makes up the cross-beta spine of amyloid-like fibrils (Nelson, et al. Nature, 2005). Each six-residue peptide not containing a proline from a putative protein sequence is threaded onto the NNQQNY structure backbone, and the energetic fit is evaluated by using the RosettaDesign program (Kuhlman et al., PNAS, 2000). To avoid problems with their disulphide bonding abilities, cysteines were substituted to serines during modeling. Additionally, the quality of the steric zipper interface is compared in terms of shape complementarity and surface area to other amyloid-like peptides reported by Sawaya et al. Nature, 2007. Based on these experimental amyloid-like peptide structures an energy threshold of -23 kcal/mol was chosen. Segments with energies equal to or below this threshold are deemed to have high fibrillation propensity.
Search & Find
We have designed this database to be intuitive and flexible. The database can be searched for a protein of interest by its name, accession identifier (GI, PDB, if applicable) or residue sequence. If we do not have an exact match for the protein, you may perform a BLAST search of our database. We welcome inquiries to have new protein sequences analyzed and added to the database (please contact us). Additionally, the database can be searched for a specific hexapeptide segments by simply entering its residue sequence. Use the advanced search to specify multiple search criteria.
Genomes, datasets, proteins or hexapeptide segments matching your query will be shown on the search results page. Click on a genome or dataset name to see the associated proteins. Click on a protein name to see its sequence, fibrillation propensity profile and segment statistics. Click on a segment sequence to see details statistics, list of proteins it is found in, and view or download the PDB model coordinates.
List grids enumerating protein and segments can be sorted ascending and descending by clicking on the column header as well as filtered by entering partial matches into the text box right below the header name. Numeric and date columns can be filtered by values such as ">0.7" or "<-23". Please note the page navigation links on the bottom of the list.
Interpreting Propensity Profile Graphs
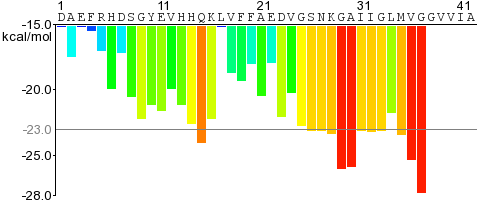 For a given protein, the energies of therein contained hexapeptides are plotted, such as for Amyloid-beta on the right.
The residue sequence is plotted along the X-axis.
Each histogram bar represent one hexapeptide starting at the indicated position in the sequence and is colored according to its Rosetta energy.
Orange-red segments with energy below the indicated energetic threshold of -23 kcal/mol (gray line), and are predicted to form fibrils.
For a given protein, the energies of therein contained hexapeptides are plotted, such as for Amyloid-beta on the right.
The residue sequence is plotted along the X-axis.
Each histogram bar represent one hexapeptide starting at the indicated position in the sequence and is colored according to its Rosetta energy.
Orange-red segments with energy below the indicated energetic threshold of -23 kcal/mol (gray line), and are predicted to form fibrils.
Hover the mouse on a histogram bar to see the sequence and energy of the corresponding segment. Click on it to see the segment's details, view the model and download its coordinates in PDB format.
Some profiles will have "blind spots" 6 residues in length or longer. Any hexapeptide that contains a proline will not be included in the profile as prolines are beta-strand breakers and incompatible with our prediction algorithm.
Peptide Segment Statistics
The following statistics are reported for each peptide segment:
- Energy: The Rosetta energy of one layer composed of two beta-strands (2 x 6 residues) in kcal/mol. Lower energies preferred.
- Shape Complementarity (Sc): The Lawrence and Colman's shape complementarity (Lawrence 1993) of the steric zipper interface (inward facing residues). Range 0.0 - 1.0; higher values preferred.
- Area of Interface: The solvent-accessible surface area (Lee 1971) at the steric zipper interface per layer in Å2. Higher values indicate more contact at the zipper interface and are preferred.
- Contact Area The contact area in Å2 between the two sheets, determined from molecular surface modeling (CCP4 sc program). Larger area preferred.
- SASA: The solvent-accessible surface area of the entire five-layer structure in Å2. Lower values indicate more compact structure and are preferred.
- Composite Score: A score taking the Rosetta energy, shape complentarity and area of the interface into account. No units; lower values preferred.
Data Download
The data for peptide segments in a specific protein or for search results can be downloaded in CSV or tab-separated text formats, and in XML format. The corresponding links are located right next to the list grid enumerating the peptide segments on the protein details page and on the search results page. We currently do not provide a download feed of the our entire database due to its size (1 GB score data plus 36 GB bzipped PDB model coordinates).